Table of Contents >> Show >> Hide
- Quick Facts (Because Your Brain Deserves a Road Map)
- What Exactly Is CJD?
- Causes of CJD (And Why “Cause” Can Mean Different Things)
- Symptoms of CJD
- Diagnosis: How Doctors Evaluate Suspected CJD
- Treatment for CJD: What Can Actually Help?
- Prognosis and Outlook
- Prevention and Safety: What You Should (and Shouldn’t) Worry About
- When to Seek Medical Care (And What to Ask)
- Experiences: What Patients, Families, and Clinicians Often Describe (A 500-Word Reality Check)
- Conclusion
Creutzfeldt-Jakob disease (CJD) is one of those conditions that sounds like it should come with a pronunciation guide and a warning labeland it does. CJD is a rare, rapidly progressive brain disease caused by prions, which are misfolded proteins that trigger other proteins to misfold too. Think of it like “bad protein origami” that spreads its terrible folding habits through the brain. The result is fast-moving damage that affects memory, behavior, movement, and eventually vital functions.
It’s also important to know two reassuring facts right up front: CJD is extremely rare, and it is not spread through casual contact like hugging, sharing a bathroom, or sitting near someone on a plane. Most families dealing with CJD are blindsided not because they “missed a risk,” but because the disease often begins subtly and can mimic other conditions.
Quick Facts (Because Your Brain Deserves a Road Map)
- What it is: A prion disease that causes rapid brain degeneration (“spongiform” changes).
- How common: About 1–2 cases per million people per year; several hundred cases annually in the U.S.
- Typical age: Most often affects older adults (commonly late 50s and up).
- Course: Often progresses over weeks to months; many people die within about a year after symptoms start.
- Treatment: No cure; care focuses on comfort, symptom relief, and safety.
What Exactly Is CJD?
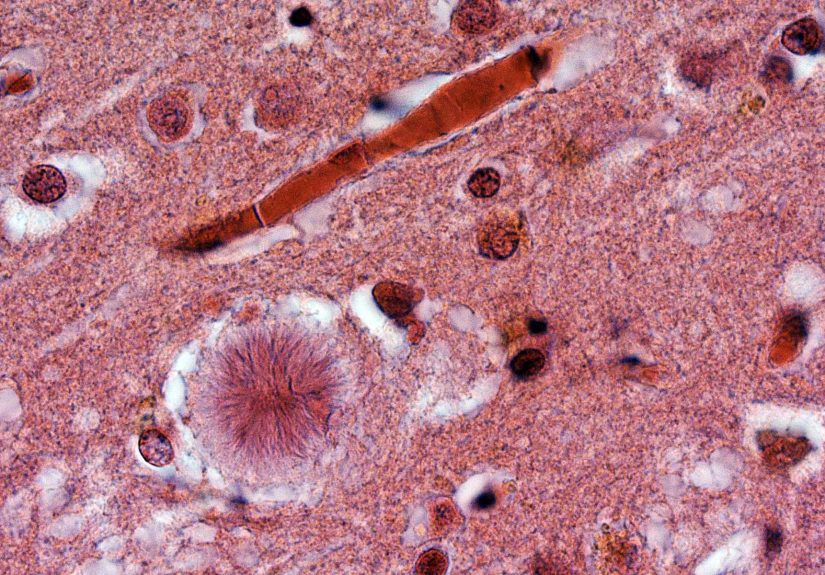
CJD is part of a family of illnesses called transmissible spongiform encephalopathies (TSEs). “Transmissible” here does not mean it spreads easily between people. It means that under very specific, unusual circumstances, prions can be transmitted (more on that later). “Spongiform” refers to the microscopic appearance of damaged brain tissue, which can look sponge-like because of tiny holes formed as brain cells are injured.
In classic CJD, the brain’s normal prion protein (a protein found naturally in the body) changes shape into an abnormal form. This abnormal form can prompt other prion proteins to misfold as well, leading to a chain reaction. Over timeoften quicklythis disrupts brain function and causes the symptoms people notice in daily life.
Causes of CJD (And Why “Cause” Can Mean Different Things)
CJD is caused by prions, but “how the prion problem begins” differs by type. Public health agencies typically describe three major categories of classic CJD: sporadic, familial (genetic), and iatrogenic (acquired in healthcare settings).
1) Sporadic CJD (Most Common)
Sporadic CJD accounts for the vast majority of cases. In this type, the misfolding of prion protein appears to occur “out of nowhere,” meaning there’s no clear exposure and no known inherited mutation. Researchers still don’t fully understand why the misfolding starts in one person and not another.
Key point: Sporadic CJD is not something you “catch” from someone, and most people with sporadic CJD have no family history of the disease.
2) Familial (Genetic) CJD
Familial CJD happens when a person inherits a mutation in the gene that encodes prion protein. This can raise the likelihood that prion proteins will misfold during the person’s lifetime. Familial prion diseases can include genetic CJD as well as related inherited prion disorders.
What this can look like in real life: Families may describe multiple relatives over generations who were diagnosed with “early dementia,” “Parkinson’s,” or “psychiatric illness,” only later learning that a genetic prion disease could have been involved. Genetic testing and counseling can be important when a familial form is suspected.
3) Iatrogenic (Rare Acquired) CJD
Iatrogenic CJD is extremely rare and refers to transmission through specific medical exposureshistorically involving contaminated biological products or instruments that contacted high-infectivity tissues (like brain tissue). Reported links have included certain historical uses of cadaver-derived human growth hormone, dura mater grafts, some corneal transplants, and, very rarely, neurosurgical instruments or specific implanted electrodes.
Modern reality: Infection-control and sterilization practices have changed substantially over time, and healthcare-associated CJD transmission is now extraordinarily uncommon.
What About Variant CJD (vCJD)?
Variant CJD (vCJD) is a separate, much rarer condition linked to exposure to bovine spongiform encephalopathy (BSE), often called “mad cow disease.” vCJD tends to affect younger people than classic CJD and is known for earlier psychiatric symptoms and painful sensory symptoms (like dysesthesia). In the U.S., reported vCJD cases have been extremely rare and are generally believed to have been acquired outside the country.
Symptoms of CJD
CJD symptoms often progress rapidly, but the first symptoms can vary. Some people begin with memory problems or confusion; others start with changes in coordination, vision, mood, or sleep. Many experts refer to CJD as a “great mimicker” because early signs can resemble more common (and sometimes treatable) conditions.
Early Symptoms (Common Starting Points)
- Changes in thinking and memory: confusion, trouble focusing, difficulty planning or following steps
- Personality or behavior changes: irritability, apathy, anxiety, depression, withdrawal
- Vision problems: blurry vision, visual disturbances, or vision loss
- Coordination issues: clumsiness, trouble walking, balance problems, dizziness-like unsteadiness
- Sleep disruption: insomnia or major changes in sleep patterns
As the Disease Progresses
As CJD advances, symptoms often become more obvious and disabling. Many people develop rapidly progressive dementia along with movement problems. A hallmark symptom is myoclonussudden, involuntary jerking movements. People may also become rigid, have speech and swallowing difficulties, and eventually enter a stage where they lose the ability to communicate and move.
A Real-World Example of Symptom Progression
Imagine a previously independent 67-year-old who begins misplacing items and struggling with routine tasks (like paying bills or following a familiar recipe). Over a few weeks, family notices unsteadiness and a “startle” responsejerky movements when surprised. Then speech becomes slower, sleep worsens, and the person seems increasingly confused. This fast timelineweeks to monthsoften pushes clinicians to consider rapidly progressive dementia causes, including prion disease, autoimmune encephalitis, infections, and metabolic disorders.
Diagnosis: How Doctors Evaluate Suspected CJD
There is no single everyday “office test” that confirms CJD with certainty during life in all cases. The only definitive confirmation historically comes from specialized testing of brain tissue (usually at autopsy; biopsy is uncommon). However, modern evaluation can strongly support a diagnosis using a combination of clinical features and specialized testing.
Why Diagnosis Can Be Tricky (But Also Urgent)
When someone has dementia that is getting worse quicklyover weeks to monthsdoctors work hard to identify treatable causes first. Some rapidly progressive dementias are reversible or partially treatable (for example, autoimmune encephalitis, certain infections, thyroid disease, severe vitamin deficiencies, or toxic exposures). A thorough workup protects patients by not missing a condition where early treatment can change outcomes.
Common Parts of a CJD Workup
- Neurologic exam and history: timing, symptom pattern, exposures, travel history, family history
- Brain MRI (especially DWI/FLAIR sequences): often one of the most helpful tools; can show patterns characteristic of CJD
- EEG: measures electrical activity; certain patterns can support diagnosis (though they are not always present)
- Spinal fluid (CSF) testing: may include biomarkers and prion-focused tests like RT-QuIC
- Bloodwork and other tests: to rule out autoimmune, infectious, metabolic, or toxic causes
- Genetic testing: considered when familial prion disease is possible
RT-QuIC: A Major Step Forward
One of the most important advances in recent years is RT-QuIC (real-time quaking-induced conversion), a test that can detect abnormal prion activity in certain samples (commonly spinal fluid) and can support diagnosis during life. Because CJD progresses quickly, having better antemortem testing can help families and care teams make decisions earlierespecially around safety, planning, and supportive care.
Ruling Out “Look-Alikes”
Specialty centers often use structured approaches to rapidly progressive dementia. Clinicians may consider categories like vascular, infectious, toxic-metabolic, autoimmune, neoplastic, iatrogenic, neurodegenerative, and systemic causes. That’s a fancy way of saying: “Let’s make sure we aren’t missing something treatable before we conclude it’s prion disease.”
Treatment for CJD: What Can Actually Help?
There is currently no cure and no proven disease-stopping therapy for CJD. Treatment focuses on comfort, symptom relief, and support for caregivers. While that can sound discouraging, supportive care can make a meaningful difference in quality of lifefor both patients and families.
Symptom Management
- Myoclonus (jerking movements): medications such as clonazepam or sodium valproate may help reduce jerks in some patients
- Pain and discomfort: treated with appropriate pain-relief strategies
- Anxiety, agitation, mood changes: may be managed with carefully chosen medications and calming routines
- Sleep problems: sleep hygiene and, when necessary, medications
- Swallowing difficulties: speech/swallow therapy guidance, texture modifications, and safety planning
- Mobility and falls: physical therapy strategies, assistive devices, home safety updates
Palliative Care and Hospice
Because CJD often progresses quickly, early involvement of palliative care can help align care with the patient’s goals and reduce distressing symptoms. Hospice can provide additional support at home or in a facility when the focus becomes comfort and dignity.
Clinical Trials and Research
Researchers continue investigating potential treatments, including approaches that aim to reduce prion protein production or interfere with misfolding. Clinical trials can be challenging because the disease is rare and fast-moving, but research is activeand families who want to explore research options can discuss referral to specialty centers or look for ongoing trials.
Prognosis and Outlook
CJD is always fatal, typically within months to about a year after symptoms begin, though timelines vary. Some people decline very rapidly, while others progress more slowly. Many patients die from complications such as infections (for example, pneumonia) that occur as swallowing, mobility, and overall resilience decline.
Although the prognosis is grim, early recognition can still matter. A faster diagnosis can prevent unnecessary procedures, reduce uncertainty, help families plan, and ensure comfort-focused care is started sooner rather than later.
Prevention and Safety: What You Should (and Shouldn’t) Worry About
Casual Contact Is Not a Risk
Everyday interactions do not spread classic CJD. Families can safely provide loving care, including touch and closeness, using standard hygiene practices.
Healthcare Settings and Instrument Safety
Prions are unusually resistant to routine sterilization, which is why medical facilities have specific protocols when prion disease is suspectedespecially for instruments that contact high-risk tissues. In some situations, the safest approach may be using specialized decontamination methods or, when practical, discarding certain instruments exposed to high-infectivity tissue. These practices are primarily relevant to surgical and procedural settings, not to routine household life.
Food and vCJD
vCJD is linked to BSE exposure, and public health measures (like surveillance and controls in food production) have greatly reduced risk. In the U.S., vCJD has remained extremely rare.
When to Seek Medical Care (And What to Ask)
If you notice a rapid decline in memory, thinking, behavior, vision, coordination, or speechespecially over weeks to monthsseek medical attention promptly. Ask for a neurological evaluation, and consider asking questions like:
- “Could this be a rapidly progressive dementia, and what treatable causes are we ruling out?”
- “Should we get an MRI with DWI and FLAIR sequences?”
- “Would spinal fluid testing, including RT-QuIC, be appropriate?”
- “Is genetic counseling or testing recommended based on our family history?”
- “Can we involve palliative care early for symptom support and planning?”
Experiences: What Patients, Families, and Clinicians Often Describe (A 500-Word Reality Check)
When people talk about CJD, the medical facts are terrifyingbut the experience is often defined by something else: speed. Families frequently describe a before-and-after line in the sand. One month a loved one is driving, joking, and living independently; the next month, everyone is arguing over whether the problem is “just stress,” a medication side effect, depression, or maybe a small stroke. That early uncertainty can be emotionally brutal, because CJD doesn’t announce itself with a neat label. It sneaks in wearing other conditions’ clothes.
Caregivers often recall the first signs as strangely ordinary: forgetting appointments, losing a familiar word mid-sentence, taking a wrong turn on a well-known route. Then come the “this is not normal” momentssudden clumsiness, unexplained falls, vision issues, or a personality shift that feels like someone turned the dimmer switch on the person you know. Some families describe their loved one becoming unusually anxious or withdrawn. Others notice insomnia that seems out of proportion to life circumstances, like the brain can’t find its off switch anymore.
One common theme is the whirlwind of appointments and tests. A primary care visit becomes a neurology referral; the neurology referral becomes imaging and spinal fluid tests. Families often say the MRI was a turning pointnot because it made things easier, but because it made things clearer. Clarity has its own weight. When doctors start using terms like “rapidly progressive dementia,” families may feel two opposite emotions at once: relief that someone is taking it seriously, and fear because the differential diagnosis can include serious conditions.
Clinicians who care for CJD patients often emphasize communication: explaining what is known, what is suspected, and what still needs to be ruled out. Families tend to value directness paired with humanityclear expectations, but not coldness. Small practical help matters a lot: guidance on fall-proofing the home, simplifying medications, preventing aspiration, and planning for 24/7 supervision when judgment and mobility decline. Many caregivers also describe the exhaustion of constant vigilance. If you’re caring for someone with suspected or confirmed CJD, you are not “dramatic” for needing helpyou are being realistic.
And here’s the part that feels almost unfairly practical: families often become project managers overnight. They gather records, track symptoms, coordinate relatives, and make decisions about feeding, hospitalizations, and comfort caresometimes faster than they ever imagined possible. People who’ve been through it frequently recommend a few coping tools: keep a simple symptom timeline, appoint one family communicator for updates, ask early about palliative care, and accept help in the form it actually arrives (yes, even if it’s a casserole). Humor sometimes shows up toonot the “this is funny” kind, but the “we’re still here” kind. In a disease that moves fast, small moments of connection can matter enormously.
Conclusion
Creutzfeldt-Jakob disease is a rare but devastating prion disorder that causes rapid neurological decline. While there’s no cure today, timely evaluation can rule out treatable look-alike conditions, and supportive care can meaningfully reduce suffering and help families plan. If rapid cognitive or movement changes appear over weeks to months, prompt medical assessmentideally with a neurology team familiar with rapidly progressive dementiacan make the path clearer, even when the outcome is difficult.